
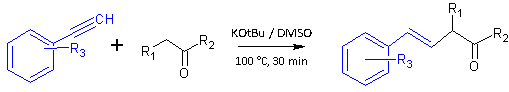
α-vinylation reaction of Trofimov et Al (1)
The authors provide a considerable list of existing routes to β,γ enones, which includes Pd-catalyzed ketone vinylations with alkenyl bromides / triflates (2), coupling reactions between α-iodoenones and alkenylzinc reagents (3) and various organobismuth, organoindium, and organostannane techniques. Many others were referenced by the authors, with this caveat: the reported reactions typically require some combination of exotic raw materials and elaborate procedures.
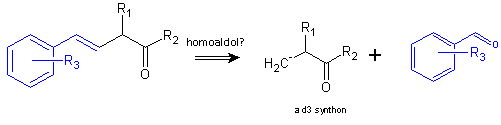
Synthetic Strategy: the homoenolate synthon (4)
Trofimov and coworkers thus provide an alternative which seems to simplify this transformation in several ways:
- Readly available starting materials are used (arylacetylenes and various types of alkyl ketones).
- Transition metal catalysis is not required.
- Absolute stereoselectivity is obtained for the (E)-alkene isomer product.
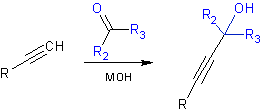
Do you remember the Favorsky Reaction? (5)
This work avoids another pitfall: the predicted Favorsky reaction product is not favored. In this reaction a ketone undergoes nucleophilic attack from an acetylide, which is formed in situ using a strong base. The Favorsky reaction is a route to propargyl alcohols and provides a protection device for terminal alkynes. By heating in KOH / IPA, the reaction can be reversed to regenerate the alkyne (5).
Something mechanistically curious is at work here. Trofimov and coworkers posit (see below) that a chelation effect is responsible for the reaction’s stereospecificity. They suggest and that a quasi-aromatic intermediate forms, which decomposes on workup. The authors further suggest that enolate additions to alkynes should produce (Z)- configured β,γ-enones, based on a predicted concerted trans-addition pathway. It would seem that the (Z)-enone that initially forms can undergo isomerization in its deprotonated form, and this is driven by participation of π electrons from the arylalkenyl group.
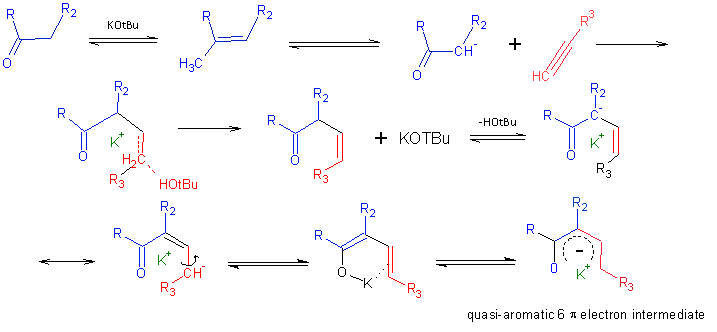
They support this proposed mechanism through experimentation, and discuss the following:
- the highest product yields are realized when an equimolar ratio of the ketone and base are used.
- Larger, more diffuse cations like Cs disturb the stereoselectivity of the reaction, suggesting a ‘templating’ capability for K cations.
- A equal product distribution of (E) and (Z) isomers at thermodynamic equilibrium is predicted by quantum mechanical calculations. The reaction is apparently under kinetic control, determined by the rate of chelation.
- At 60°C, a 3:2 isomeric mixture was observed. Trofimov and coworkers attribute this loss of selectivity to incomplete chelation at this lower temperature.
The authors provide a wealth of examples, including dialkyl-, alkylaryl-, and alkyl heteroaryl ketone starting materials. Alkyne partners were all arylacetylene compounds. Screening studies suggested that DMSO is perhaps uniquely suited for this reaction; Other dipolar aprotic solvents (NMP, DMF) as well as THF were utter failures.
Careful evaluation led to the selection of potassium t-butoxide over alkali metal hydroxide based. The Trofimov Group and other researcher have worked to develop the utility of this so-called ‘DMSO Superbase’ system. Reaction yields under their optimized conditions in the present work were between 61-92%.
Their general procedure was described as follows:
Ketone (4 mmol), phenylacetylene (4 mmol), potassium t-butoxide (4 mmol) and DMSO (10 mL) were heated at 100°C for 30 minutes, when the reaction was judged complete. The crude reaction mixture was cooled to room temperature and then diluted with water (10 mL). This was extracted with ether (10 mL x 4) and the organic extract was washed with water (5 mL x 3) and dried over MgSO4. The dried extract phase was concentrated in vacuo and the residue purified by silica gel chromatography (hexane / benzene, 1:0 to 0:1 gradient).
Artie McKim
References
(1) Trofimov, B.A.; Schmidt, E.Y; Zorina, N. V.; Ivanova, E. V.; Usahkov, I. A. J. Org. Chem. 2012, 77, 6880-6886.
(2) Chieffi, A.; Kamikawa, K.; Ahman, J.; Fox, J.M.; Buchwald, S.L. Org Lett. 2001, 3, 1897.
(3) Negishi, E.; Owezarczyk, Z.R.; Swanson, D.R. Tetrahedron Lett. 2000, 125, 10494.
(4) Wyatt, P.; Warren, S. Organic Synthesis: Strategy and Control J.W. Wiley Pubs. (2007) p 194.
(5) Greene, T.; Wuts, P. Protective groups in organic synthesis, Wiley-Interscience, 1998. Propargyl alcohols containing protons α to the hydroxy group may undergo further rearrangement to provide α,β-unsaturated ketones; this is known as theMeyer-Schuster Rearrangement when the process is acid catalyzed. See also the Rupe Rearrangement of tertiary alcohols containing an α-acetylenic group
(6) Y. Yuan, I. Thome, S.-H. Kim, D. Chen, A. Beyer, J. Bonnamour, E. Zuidema, S. Chang & C. Bolm, Adv. Synth. Catal. 2010, 352, 2892-2898 b) B.A. Trofimov, E.Y. Schmidt, N.V. Zorina, E.V. Skital’tseva & A.I. Mikhaleva, Chemistry of Heterocyclic Compounds 2010, 46(5), 623-624