
Sulfide Oxidation Method Evaluation
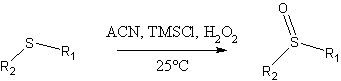
The oxidation procedure of Bahrami, Khodaei and coworkers (1)
Some features of this method:
- fast reaction times: typical examples provided were driven to completion within minutes!
- High yields for the examples provided (>92%), with negligible amounts of sulfone byproduct. As an example, a model experiment reported by the authors using dimethyl sulfide as starting material produced a 95% isolated yield of DMSO.
- Simple workup.
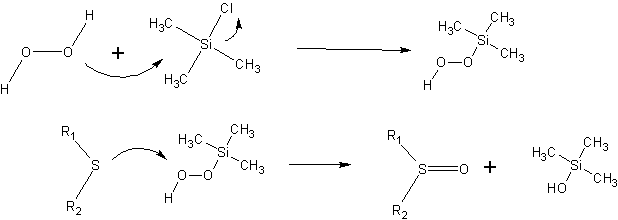
Activation of hydrogen peroxide by TMSCl in a sulfide oxidation procedure.
The authors provide a plausible mechanistic explanation of how hydrogen peroxide is activated. Nucleophilic attack on TMSCl by peroxide produces an intermediate which is capable of oxidizing dialkylsulfides.
Trimethylsilylalcohol is produced as a byproduct; the authors state that this is easily removed during workup due to its water solubility.
We set up some small test reactions to evaluate the method to make our compounds. After a little trial-and-error, a few things became apparent:
- In our case, a slight excess of hydrogen peroxide (about 1.06 molar ratio relative to sulfide starting material) seemed to work best. The authors (1) recommended a twofold excess of peroxide; with our compounds we experienced overoxidation resulting in sulfone byproduct formation.
- A slightly longer reaction time than those described in the paper was preferred in our experience. We allowed the mixture to stir for about 40 minutes after peroxide addition was complete, compared to the very brief reaction times (<10 minutes)described by the authors. We weren’t in such a hurry and the reaction smoothly ran to completion in this time frame.
- We modified the author’s workup procedure to minimize the amount of water used during the quench step and subsequent extraction. The published workup used a volume of water roughly equal to that of the reaction mixture. As the desired sulfoxide compounds have appreciable water solubility, we cut the water charge to the reactor back to about 1/3rd of the reaction mixture volume. This was just enough to cause phase separation between the ethyl acetate (and acetonitrile) layer and the aqueous phase.
- We later found that reducing the water used during extraction left a considerable amount of what we thought to be acetic acid in the organic phase. This complicated distillation on a small scale and we chose to neutralize the water phase with 10% NaOH. An equivalent of NaOH for each equivalent of peroxide charged in the reaction solved this problem.
Ultimately, our adapted procedure looked like this:
To a three necked 3L RBF equipped with mechanical stirring and a pressure equalizing dropping funnel was charged acetonitrile (1000 ml), dialkylsulfide (0.192 mol, MR 1) and trimethylsilylchloride (0.210 mol, 22.9 g, MR 1.09 MR). To the dropping funnel were charged 50% aqueous hydrogen peroxide (13.74g solution, 0.202 mol, 1.05 MR) diluted (3) with water (10.5 g).
The peroxide solution was added dropwise over 20 minutes with vigorous stirring. Mild warming occurs and the reaction is left to stir an additional 40 minutes. An aliquot was withdrawn for GC analysis (2) to assure the absence of starting material.
Water is added to quench the reaction, followed by EtOAc (500 mL). The pH of the aqueous phase is adjusted to > 7 using 10% NaOH (about 100 mL was used). The organic phase was separated and the aqueous phase was extracted further with EtOAc (3 x 500 mL). The combined organic phases were combined, dried over MgSO4, filtered and concentrated on the rotovap. The resulting residue is essentially pure product, yielding about 85%. In our case, the product was purifed by distillation under reduced pressure.
In closing the method of Bahrami, Khodaei, and coworkers was ideal for our purposes. It was effectively scaled to produce about 50g crude material and could seemingly be scaled further. If we have the need to do this, a factors worth optimizing:
- we would consider the actual dilution requirements of the reaction as acetonitrile consumption is inefficient when scaled directly from the published procedure.
- On a production scale you would likely boil away much of the acetonitrile reaction solvent before EtOAc (or appropriate extarction solvent) addition. A step to test for unconsumed peroxide would be a good idea.
Artie McKim.
(1) Bahrami, K.; Khodaei, M.M.; Yousefi, B.; Arabi, M.S. Tetrahedron Letters 51 (2010) 6939-6941.
(2) We found it difficult to track the reaction’s progress by TLC as the target compounds weren’t easily visualized using UV detection or in an iodine chamber. Vanillin stain works reasonably well, showing the compound as a ‘white’ spot. Methanol (10%) in DCM worked as the (silica) TLC mobile phase for our compounds.
(3) We diluted the hydrogen peroxide down to about 30% to match the prescription of the Bahrami, Khodaei paper. This probably isn’t required if the addition rate is well controlled.