
A recent report (1) from workers at Chonnam National University (Gwangju, Korea) describes a benzimidazole synthesis which:
- produces good product yields (40-98%, for about 30 examples)
- and proceeds in one pot from three readily available components: sodium azide, an aldehyde, and 2-haloanilines
- shows good functional group tolerance(nitro-, ester-, chloro-, and various heterocyclic functionalities on the aldehyde or haloaniline component).
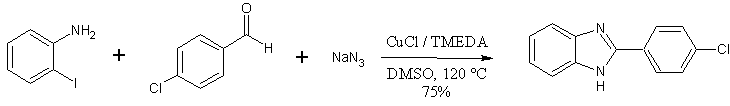
The Benzimidazole Synthesis of Lee and coworkers (1)
Naturally, there are many established ways to synthesize benzimidazoles, which are important substances used in the design of bioactive substances (2). Recent work has sought to address specific drawbacks associated with these methods, which can include harsh reaction conditions and complicated product mixtures.
Further developments have focused on the use of 2-haloacetanilides, 2-haloarylamidines, arylamino oximes, and N-arylbenzimidamides (3). This work notable due to the useful anthelmintic properties. Anthelmintic agents work to kill or repel intestinal worms. A review (3) discusses the synthesis of benzimidazoles, and cites the breakthrough discovery of thiabendazole by researchers at Merck in 1961. Thiabendazole was found to have potent broad spectrum activity against gastrointestinal parasites.
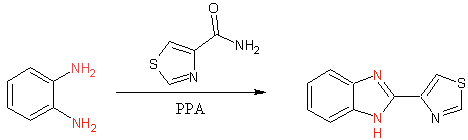
Early thiabendazole synthesis (3)
The initial synthesis of thiabendazole occured via dehydrative cyclization of 1,2 diaminobenzenze in polyphosphoric acid (PPA). The commercialized process involved the conversion of N-arylamidines using hypochlorite (4). Although this process can be performed in ‘one-pot’ fashion it is more typically performed in two steps.
The ‘one-pot’ benzimidazole synthesis described by Lee et. Al. is showcased by its ability to produce thiabendazole in one step, from readily available starting materials (2-haloanilines, thiazole-4-carboxaldehyde) – in 97% yield.
Their work builds on the report of Driver and coworkers (5) that showed that benzimidazoles could be had from 2-azidoanilines in good yield. Indeed, Lee proposes a mechanism that produces an azidoaldimine intermediate, which foregoes the multistep preparation of 2-azidoaniline starting materials.
One proposed mechanistic pathway is shown, with the following steps:
- initial in situ formation of an aldimine, via addition of aniline to an aldehyde;
- Ar-X insertion of the copper catalyst;
- Cu-azide association, with transfer of azide to the aromatic ring;
- loss of nitrogen with concomitant ring formation, and catalyst regeneration
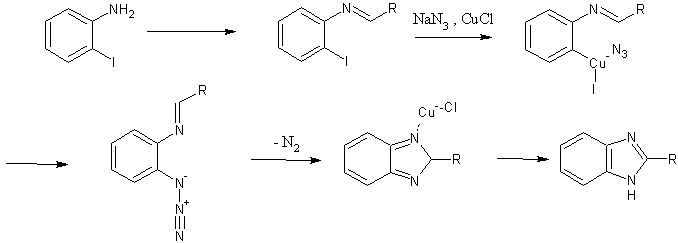
One mechanistic explanation proposed by Lee and coworkers (1).
In developing their method, they investigated a number of factors:
- Solvent. DMSO outperformed other polar solvents (NMP, DMF, DMAc). Less polar solvents failed (toluene, diglyme).
- Source of Copper catalyst. The oxidation state of copper was not a factor, as Cu(I) and Cu(II) salts showed similar performance.
- Ligand Evaluation. Ligand selection was not a large factor. Several were tested; ultimately TMEDA was selected.
- Substituents on the aniline / pyridyl component. Base sensitive substituents were tolerated (benzoate ester) and 3-Cl groups were fine. The sensitivity to a broad range of substituents (the usual EWD- and ED-groups) was not rigorously determined
- Nature of the haloaniline. Although both bromo- and iodoaniline examples were given, the predominance of iodoaniline examples suggests it was prefered by the authors for unstated reasons.
- Reactivity of various aldehyde reactants. Aldehydes of varying classes were evaluated. Yields from aromatic substrates bearing ED groups(benzaldehyde, 4-Cl benzaldehyde, 4-methoxybenzaldehyde) produced the highest product yields. Aliphatic aldehydes produced noticeably lower yields, with the curious exception of pivaldehyde. Several heterocyclic aldehydes (2- furyl- and 2-thionylaldehyde were tested and provided good results.
A synopsis of the Lee Procedure follows:
CuCl (0.1 mmol), haloaniline (2.0 mmol), TMEDA (0.1 mmol), NaN3 (4.0 mmol), aldehyde (2.4 mmol) were combined in DMSO mL), The mixture was heated at 120 C for 12 hours. After cooling to room temperature the mixture was poured onto EtOAc (50 mL), washed with brine (25 mL) and water (25 mL). The organic phase was dried over Mg2SO4, and the residue from evaporation was purified by column chromatography (1:1 hexane / EtOAc mobile phase).
Artie McKim.
References
(1) Kim, Y.; Kumar, M.R.; Park, N.; Heo, Y.; Lee, S. J. Org. Chem. 2011, 76, 9577-9583.
(2) Tumulty, D.; Cao, K.; Homes, C.P. Org Lett. 2001, 3, 83.; Wu, Z. Rea. P.; Wickham, G.; Tetrahedron Lett. 2000, 41, 9871.; Chari, M.A.; Shobha, P.S.D.; Mukkanti, K. J. Heterocycl. Chem. 2010, 47, 153.
(3) Townsend, L.B.; Wise, D.S. Parasitology Today 6, 4 (1990) 107-112.
(4) Grenda, V. J.; Jones, R.E; Gal,G.; Sletzinger J. Org Chem. 30 (1965), 259-261.
(5) Shen, M.; Driver, T.G. Org Lett. 2008, 10, 3367.